Luật định về thiết bị y tế của EU (MDR) số 2017/745

|
Luật (EU) 2020/561 của Nghị viện và Hội đồng châu Âu ngày 23 tháng 4 năm 2020 sửa đổi Luật (EU) 2017/745 đối với các thiết bị y tế được công bố vào ngày 24 tháng 4 năm 2020 trên Tạp chí chính thức của Nghị viện châu Âu! Mục tiêu chính của việc sửa đổi là hoãn ngày nộp đơn từ ngày 26 tháng 5 năm 2020 đến ngày 26 tháng 5 năm 2021. Cùng với việc hoãn này, các ngày áp dụng những điều khoản khác cũng đã được thông qua. Xem thông tin chính thức của Luật định sửa đổi tại đây: https://eur-lex.europa.eu/eli/reg/2020/561/oj |
|---|

Khởi đầu thuận lợi cho việc tuân thủ Luật định về thiết bị y tế của EU

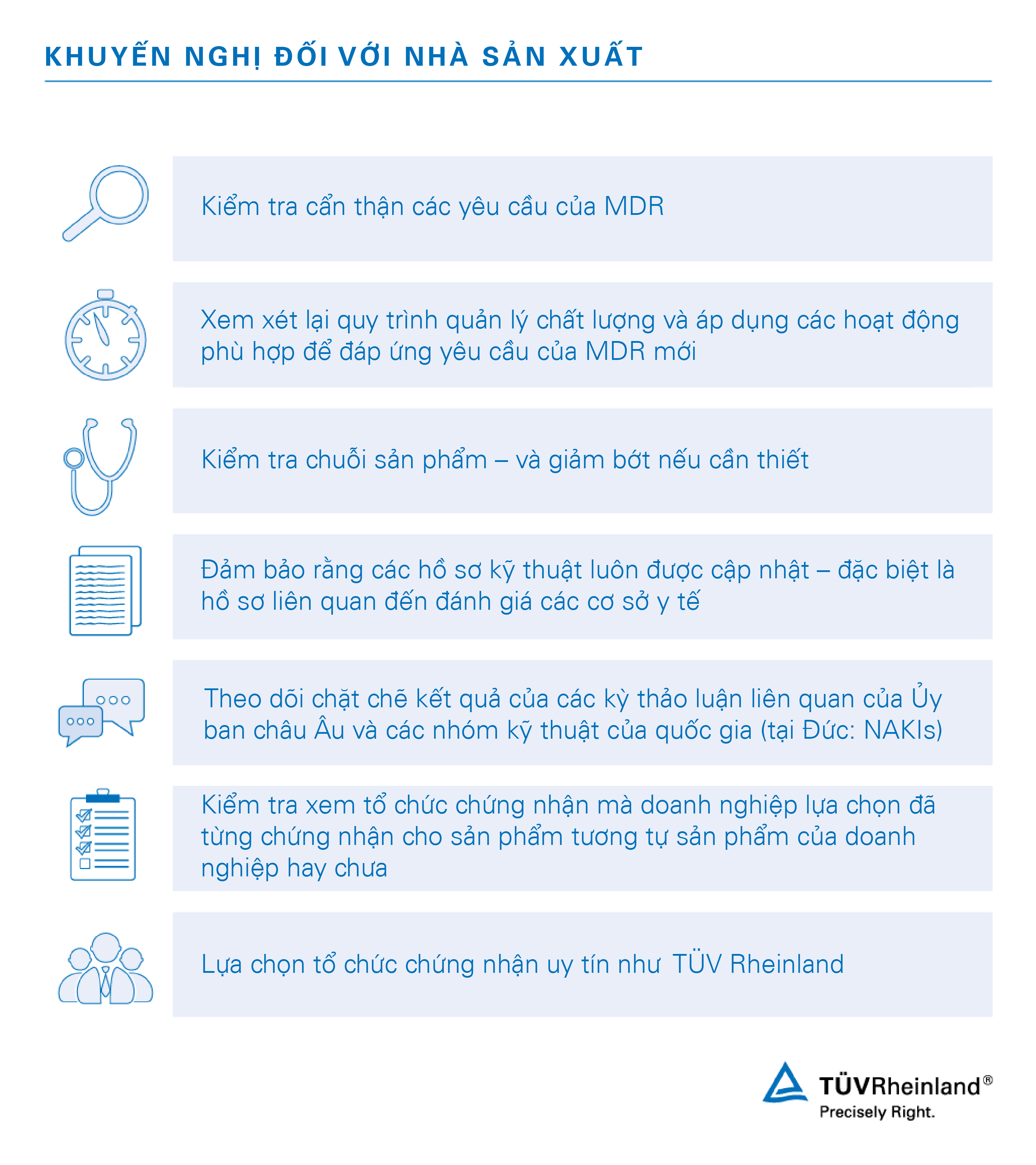
Các nhà sản xuất thiết bị y tế đang phải đối mặt với những yêu cầu mới của Luật định MDR 2017/745 có hiệu lực vào ngày 25/5/2017 và áp dụng vào ngày 26/5/2020.
Chúng tôi được đánh giá cao trong lĩnh vực thiết bị y tế bởi sự uy tín cùng chuyên môn sâu rộng. Với tư cách là Tổ chức chứng nhận lâu năm, chúng tôi luôn chủ động xác minh tính tuân thủ theo Chỉ thị về thiết bị y tế số 93/42/EEC (MDD) và Chỉ thị về thiết bị y tế có thể cấy ghép active số 90/385/EEC (AIMDD), đều sẽ được thay thế bởi luật định mới. Năng lực và khả năng của chúng tôi trong lĩnh vực này đã được khẳng định và chúng tôi muốn chia sẻ những hiểu biết của mình với các khách hàng hiện tại cũng như đối tác mới.
Với kinh nghiệm và vị thế vững chắc trong vai trò là Tổ chức chứng nhận theo luật định về thiết bị y tế hiện hành, chúng tôi hoàn toàn đủ khả năng hỗ trợ doanh nghiệp trong việc chuyển đổi sang quy định mới
Liên hệ chúng tôi để tìm hiểu thêm về yêu cầu cũng như lộ trình áp dụng của MDR 2017/745 và những ảnh hưởng đến doanh nghiệp
Đảm bảo khả năng tiếp cận thị trường EU với các đánh giá MDR
Với việc Luật định MDR 2017/745 thay thế các quy tắc quản lý thiết bị y tế hiện hành tại châu Âu, nhà sản xuất sẽ sớm phải tiến hành đánh giá lại sản phẩm của mình để đảm bảo tính tuân thủ. Quy trình đánh giá sự phù hợp với MDR cung cấp cho doanh nghiệp cơ hội đạt được chứng nhận cần thiết để bán sản phẩm trên thị trường châu Âu. Đạt được tính tuân thủ, điều kiện tiên quyết để thành công về mặt pháp luật.
Trong quá trình doanh nghiệp thực hiện tuân thủ, chúng tôi có thể hỗ trợ thông tin, thông báo và sau cùng là tiến hành xem xét cũng như đánh giá các tài liệu kỹ thuật cần thiết. Với mạng lưới chuyên gia giàu kinh nghiệm trong ngành trên toàn cầu, chúng tôi cung cấp các dịch vụ liên quan đến thiết bị y tế một cách toàn diện.
Đánh giá sự phù hợp của các thiết bị y tế
Hiện nay các dịch vụ của chúng tôi tập trung vào giai đoạn chuyển đổi và lộ trình để tuân thủ MDR 2017/745. Đội ngũ chuyên gia của chúng tôi có thể hỗ trợ doanh nghiệp đáp ứng đúng thời hạn cũng như giải quyết mọi vấn đề phát sinh trong quá trình đưa các thiết bị y tế vào thị trường châu Âu.
Đối tác tin cậy của doanh nghiệp để đảm bảo tuân thủ chỉ thị về thiết bị y tế
Là tổ chức thử nghiệm và cấp chứng nhận cũng như chuyên gia trong lĩnh vực tiếp cận thị trường toàn cầu, chúng tôi cung cấp các dịch vụ toàn diện trong ngành thiết bị y tế mà tất cả các giai đoạn thực hiện đều từ một nguồn. Bên cạnh việc hỗ trợ doanh nghiệp định hướng chuyển đổi nhằm tuân thủ Quy định mới về thiết bị y tế của châu Âu, chúng tôi còn cung cấp những dịch vụ như đánh giá hệ thống quản lý chất lượng dành cho các nhà sản xuất, nhà cung ứng và đại lý bán hàng thiết bị y tế và thử nghiệm thiết bị y tế. Chúng tôi luôn đổi mới và sẵn sàng cho các xu hướng tương lai trong lĩnh vực số hóa như kết nối không dây, y học từ xa, ứng dụng y tế, bảo mật mạng, bảo vệ dữ liệu cá nhân, v.v.
Trao đổi với chuyên gia để thực hiện tuân thủ MDR ngay hôm nay.
Hỏi đáp về Quy định mới của châu Âu đối với thiết bị y tế (MDR 2017/745)
Tài liệu tham khảo
| Thông tin quan trọng về sửa đổi MDR | 280 KB | Tải xuống |
Liên hệ

/tuv-rheinland-medical-device-single-audit-program-mdsap-ad-106934074_core_4_3.jpg)

/tuv-rheinland-de19_p05_ivd09-lp_core_4_3.jpg)
{kind=link}