

Droga do sukcesu z naszym kompleksowym portfolio globalnych usług badań i audytu
Wyroby medyczne są wykorzystywane do wielu różnych celów, od leczenia drobnych ran po ratowanie życia. Na poziomie międzynarodowym i krajowym ustanowiono złożony system zasad i przepisów w celu zapewnienia bezpieczeństwa urządzeń. Producenci mogą mieć trudności z monitorowaniem przepisów obowiązujących w poszczególnych krajach i tym samym wątpliwości w odniesieniu do zgodności ich produktów z wymaganiami rynku docelowego.
Jako jednostka certyfikująca i badająca wyroby medyczne, o wysokim poziomie wiedzy specjalistycznej w tej dziedzinie, oferujemy szeroki zakres usług zapewniających zgodność produktów na rynku globalnym. Usługi te obejmują:
Dopuszczenia międzynarodowe dla wyrobów medycznych (international approval medical) specjalnie dostosowane do wymagań rynków w Azji, Australii, Europie, Kanadzie, USA i regionie Ameryki Południowej.
Zatwierdzenia dla Europy zgodnie z nowym rozporządzeniem w sprawie wyrobów medycznych 2017/745 (MDR) i europejskim rozporządzeniem w sprawie wyrobów medycznych do diagnostyki in vitro 2017/746 (IVDR).
Nowość: zatwierdzenia dla Wielkiej Brytanii (UK MDR): TÜV Rheinland UK Ltd. jest teraz wyznaczona jako upoważniony brytyjski organ (UK Approved Body) zgodnie z brytyjskimi przepisami dotyczącymi wyrobów medycznych z 2002 r. (SI 2002 nr 618, z późniejszymi zmianami) (UK MDR)
Nasi eksperci posiadają bogate doświadczenie w zakresie wymogów obowiązujących norm, rozporządzeń i przepisów oraz wspierają klientów w uzyskiwaniu dostępu do rynków docelowych. Nasi eksperci, audytorzy i laboratoria testowe są do Twojej dyspozycji - lokalnie na całym świecie.
Skontaktuj się z nami, aby uzyskać szczegółowe informacje na temat naszych usług związanych z wyrobami medycznymi.
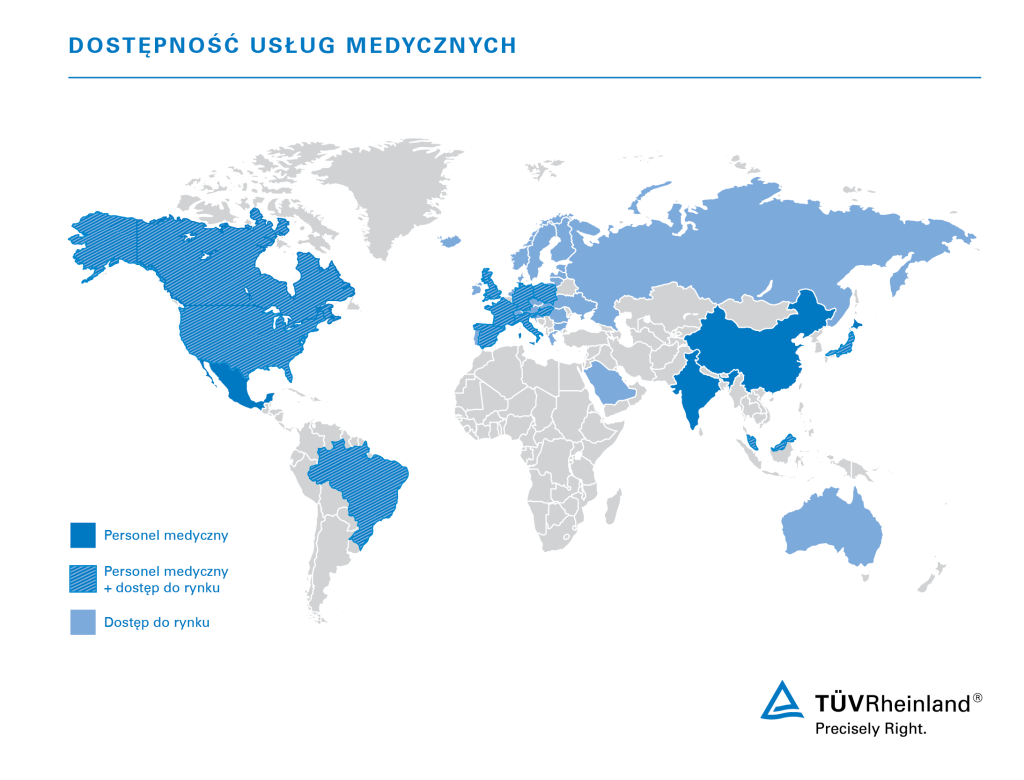
Dzięki własnym laboratoriom, ekspertom i audytorom, TÜV Rheinland działa globalnie i jest blisko klientów na całym świecie.
Nowe przepisy europejskie stanowią wyzwanie dla producentów
Jednym z największych wyzwań stojących obecnie przed producentami i/lub dystrybutorami wyrobów medycznych jest nowe rozporządzenie w sprawie wyrobów medycznych (MDR) oraz nowe rozporządzenie w sprawie diagnostyki in vitro (IVDR). Zastępują one poprzednie dyrektywy MDD (93/42/EWG), AIMD (90/385/EWG) i IVDD (98/79/WE).
W Europie nowe dyrektywy MDR i IVDR powodują niepewność w odniesieniu do certyfikacji wyrobów medycznych, w szczególności ze względu na krótkie ramy czasowe. Producenci stają przed wyzwaniem zweryfikowania ważności wszystkich istniejących deklaracji zgodności, a teraz dodatkowo muszą przeprowadzić certyfikację produktów, które wcześniej nie wchodziły w zakres dyrektyw dotyczących wyrobów medycznych.
Dopuszczenia międzynarodowe dla wyrobów medycznych - UKCA: dostęp do Wielkiej Brytanii

Po opuszczeniu Unii Europejskiej Wielka Brytania wprowadziła Rozporządzenie dotyczące wyrobów medycznych z 2002 r. (SI 2002 nr 618, z późniejszymi zmianami) (UK MDR) określające zasady wprowadzania wyrobów medycznych i wyrobów medycznych do diagnostyki in vitro na rynek brytyjski.
Aby osiągnąć płynne wejście na rynek urządzeń medycznych wyższej klasy, konieczna jest współpraca z brytyjskim zatwierdzonym organem. Ponadto należy polegać na partnerze, który rozumie branżę i ma bogate doświadczenie w zakresie testowania i certyfikacji produktu pod kątem gotowości rynkowej.
TÜV Rheinland UK Ltd. jest brytyjską jednostką zatwierdzoną zgodnie z brytyjskim rozporządzeniem MDR i posiada wiedzę specjalistyczną potrzebną do wejścia na rynek. Zapewniamy kompleksowe usługi w zakresie badań i certyfikacji z jednego źródła.
Znak UKCA
Na chwilę obecną producenci mogą nadal wprowadzać na rynek brytyjski swoje wyroby medyczne, wyroby do diagnostyki in vitro lub aktywne wyroby medyczne do implantacji ze znakiem CE (posiadające certyfikat medyczny CE). W czerwcu 2023 r. rząd Wielkiej Brytanii przedłużył okres przejściowy dla wyrobów medycznych w zależności od ich klasyfikacji. Zdecydowanie zalecamy jednak ubieganie się o certyfikację UKCA, aby uniknąć opóźnień pod koniec okresu przejściowego.
Dzięki znakowi UKCA można zapewnić, że wyrób medyczny spełnia wymogi brytyjskiego rozporządzenia MDR. Aby określić obowiązujące wymagania, należy najpierw sklasyfikować wyrób i wybrać odpowiednią procedurę oceny zgodności, aby później móc otrzymać certyfikat medyczny CE.

